Bioinformatics data management and epigenome analysis methods
Nathan Sheffield, PhDoutline
Motivation and background
Epigenome analysis
|
|
20%
50%
30%
|
Data management
◁ Questions ▷
Most pipelines require individual metadata organization


What if?


Why is this hard to do?
Because of microwave syndrome....
Because of microwave syndrome....
Microwave syndrome



In user interface design, prioritizing easy access to integrated functions over their individual components.




The UNIX philosophy

[T]he power of a system comes more from the relationships among programs than from the programs themselves.
Many UNIX programs do quite trivial tasks in isolation, but, combined with other programs, become general and useful tools.
- Kernighan and Pike, The UNIX Programming Environment (1983, p. viii)
Many UNIX programs do quite trivial tasks in isolation, but, combined with other programs, become general and useful tools.
- Kernighan and Pike, The UNIX Programming Environment (1983, p. viii)







Problem
Solution


PEP: Portable Encapsulated Projects

 PEP format
PEP format
sample_name,protocol,organism,input_file
frog_0h,RNA-seq,frog,/path/to/frog0.gz
frog_1h,RNA-seq,frog,/path/to/frog1.gz
frog_2h,RNA-seq,frog,/path/to/frog2.gz
frog_3h,RNA-seq,frog,/path/to/frog3.gz
PEP format
sample_name,protocol,organism,input_file
frog_0h,RNA-seq,frog,/path/to/frog0.gz
frog_1h,RNA-seq,frog,/path/to/frog1.gz
frog_2h,RNA-seq,frog,/path/to/frog2.gz
frog_3h,RNA-seq,frog,/path/to/frog3.gz
sample_table: /path/to/samples.csv
output_dir: /path/to/output/folder
other_variable: value
Add programmatic sample and project modifiers.
Automatically build new sample attributes from existing attributes.
Without derived attribute:
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | /path/to/frog0.gz |
| frog_1h | 1 | RNA-seq | frog | /path/to/frog1.gz |
| frog_2h | 2 | RNA-seq | frog | /path/to/frog2.gz |
| frog_3h | 3 | RNA-seq | frog | /path/to/frog3.gz |
Using derived attribute:
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | my_samples |
| frog_1h | 1 | RNA-seq | frog | my_samples |
| frog_2h | 2 | RNA-seq | frog | my_samples |
| frog_3h | 3 | RNA-seq | frog | my_samples |
| crab_0h | 0 | RNA-seq | crab | your_samples |
| crab_3h | 3 | RNA-seq | crab | your_samples |
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | my_samples |
| frog_1h | 1 | RNA-seq | frog | my_samples |
| frog_2h | 2 | RNA-seq | frog | my_samples |
| frog_3h | 3 | RNA-seq | frog | my_samples |
| crab_0h | 0 | RNA-seq | crab | your_samples |
| crab_3h | 3 | RNA-seq | crab | your_samples |
Project config file:
sample_modifiers:
derive:
attributes: [input_file]
sources:
my_samples: "/path/to/my/samples/{organism}_{t}h.gz"
your_samples: "/path/to/your/samples/{organism}_{t}h.gz"Benefit: Enables distributed files, portability
Add new sample attributes conditioned on values of existing attributes
Before:
| sample_name | protocol | organism |
| ------------- | :-------------: | -------- |
| human_1 | RNA-seq | human |
| human_2 | RNA-seq | human |
| human_3 | RNA-seq | human |
| mouse_1 | RNA-seq | mouse |
After:
| sample_name | protocol | organism | genome |
| ------------- | :-------------: | -------- | ------ |
| human_1 | RNA-seq | human | hg38 |
| human_2 | RNA-seq | human | hg38 |
| human_3 | RNA-seq | human | hg38 |
| mouse_1 | RNA-seq | mouse | mm10 |
| sample_name | protocol | organism |
| ------------- | :-------------: | -------- |
| human_1 | RNA-seq | human |
| human_2 | RNA-seq | human |
| human_3 | RNA-seq | human |
| mouse_1 | RNA-seq | mouse |
Project config file:
sample_modifiers:
imply:
- if:
organism: human
then:
genome: hg38
- if:
organism: mouse
then:
genome: mm10Benefit: Divides project from sample metadata
Define activatable project attributes.
project_modifiers:
amendments:
diverse:
metadata:
sample_annotation: psa_rrbs_diverse.csv
cancer:
metadata:
sample_annotation: psa_rrbs_intracancer.csvBenefit: Defines multiple similar projects in a single file
Looper
Deploys pipelines across samples by connectingsamples to any command-line tool

protocol_mappings:
RNA-seq: rna-seq
pipelines:
rna-seq:
name: RNA-seq_pipeline
path: path/to/rna-seq.py
arguments:
"--option1": sample_attribute
"--option2": sample_attribute2Looper features
Run your entire project with one line:
looper run project_config.yamlprotocol_mappings:
RRBS: rrbs
WGBS: wgbs
EG: wgbs.py
SMART-seq: rnaBitSeq -f; rnaTopHat -f
ATAC-SEQ: atacseq
DNase-seq: atacseq
CHIP-SEQ: chipseqpipeline_key:
name: pipeline_name
arguments:
"--option" : value
resources:
default:
file_size: "0"
cores: "2"
mem: "6000"
time: "01:00:00"
large_input:
file_size: "2000"
cores: "4"
mem: "12000"
time: "08:00:00"compute:
slurm:
submission_template: templates/slurm_template.sub
submission_command: sbatch
localhost:
submission_template: templates/localhost_template.sub
submission_command: shAdjust compute package on-the-fly:
> looper run project_config.yaml --compute localhostLooper only submits jobs for samples not already flagged as running, completed, or failed.
looper check project_config.yamllooper summarize project_config.yaml
Comparison

Prealignments
Nuclear-mitochondrial DNA (NuMts) confuse aligners







Output

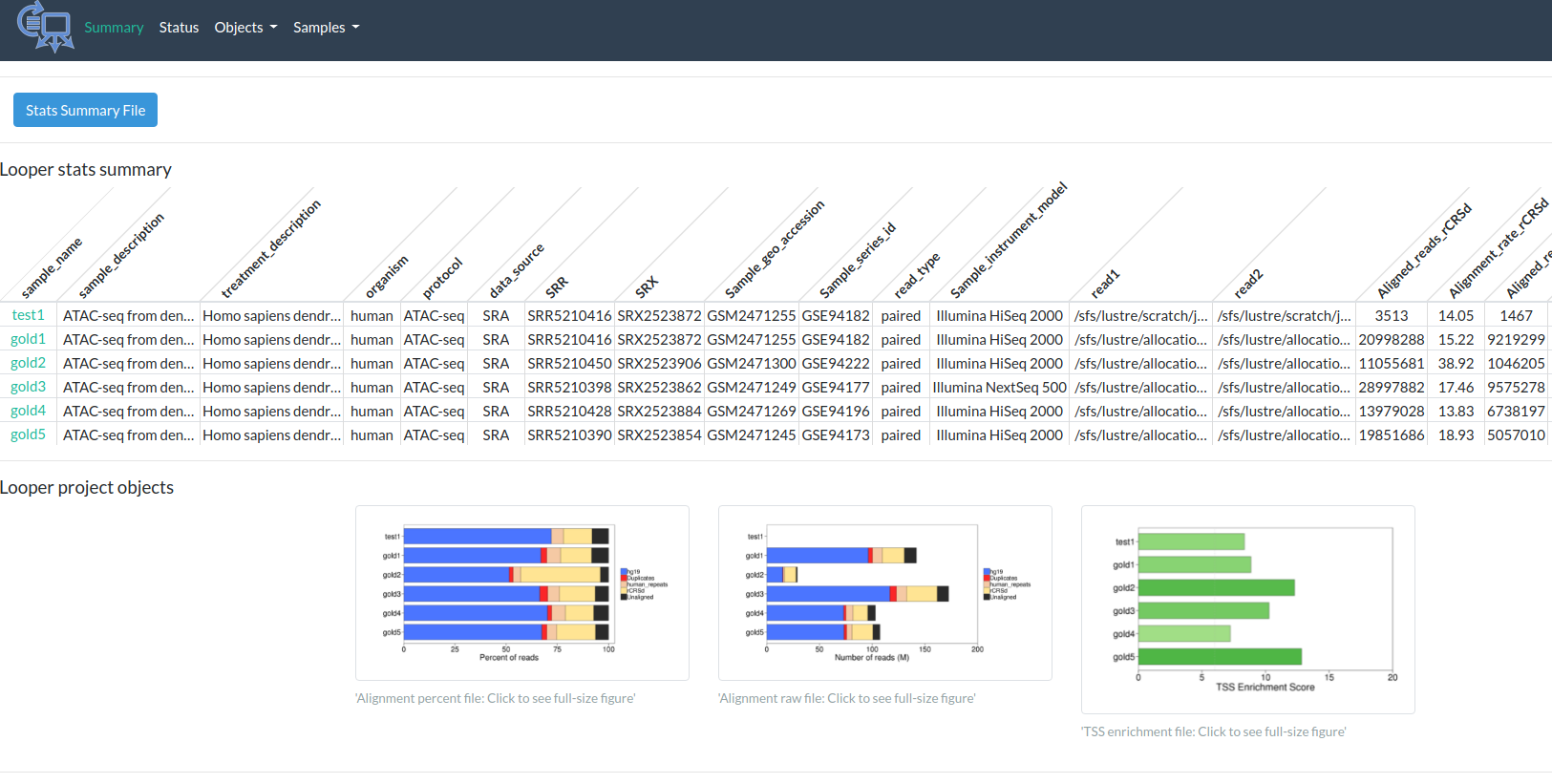
http://code.databio.org/PEPATAC/files/examples/gold/summary.html

Questions
Epigenome analysis methods
LOLA
Locus Overlap Analysis
Locus Overlap Analysis
MIRA
Methylation-based Inference of Regulatory Activity
Methylation-based Inference of Regulatory Activity
Locus Overlap Analysis












A shiny app and server for interactive LOLA analysis.
Public server: http://lolaweb.databio.org
GitHub: https://github.com/databio/LOLAweb
Public server: http://lolaweb.databio.org
GitHub: https://github.com/databio/LOLAweb
DEMO
Methylation-based Inference of Regulatory Activity (MIRA)
DNA methylation

DNA methylation

 Bisulfite-seq
Bisulfite-seq

 Region pooling
Region pooling









Thank You
Sheffield lab
John Lawson
Vince Reuter
Ognen Duzlevski
Jason Smith
Jianglin Feng
Michal Stolarczyk
Aaron Gu
Anant Tewari
Christoph Bock
Andre Rendeiro
Johanna Klughammer
Howard Chang
Ryan Corces
Yuning Wei
Jin Xu
Funding:


