Choose your own adventure through Sheffield lab research
Nathan Sheffield, PhDMission statement
We develop and apply computational methodsto organize, analyze, and understand large epigenomic data.

Biological motivation

Cells alter phenotype by using DNA differently.

Breakdowns lead to disease
Full-stack bioinformatics

Full-stack bioinformatics


Full-stack bioinformatics

Full-stack bioinformatics

Augmented Interval List

Full-stack bioinformatics


Full-stack bioinformatics

Analysis of DNA methylation in Ewing sarcoma

Full-stack bioinformatics


Bonus (unpublished) topics
- [bedbase.org](http://bedbase.org)Locus Overlap Analysis












Methylation-based Inference of Regulatory Activity (MIRA)






Coordinate Covariation Analysis (COCOA)
Goal: understand variation among individuals

Supervised differential analysis

Supervised continuous analysis

Unsupervised analysis
Epigenomic data: high-dimensional
and low-interpretable

Dimensionality reduction

Even with known groups
How can we annotate the source of variation?
COCOA Overview
 John Lawson
John Lawson
Coordinate Covariation Analysis
- Quantify variation into a 'target variable'
- Supervised (e.g. clincial variable).
- Unsupervised (e.g. PCA)
- Annotate target variable with region sets.
What is epigenetic signal covariation?
What is epigenetic signal covariation?

What is epigenetic signal covariation?

What is epigenetic signal covariation?

What is epigenetic signal covariation?

Covariation informs source of observed variation
1. Choose target variable
What is the variation we'd like to explain?
Supervised target

Unsupervised target

2. Quantify correlation with target variable

2. Quantify correlation with target variable

Permutation tests establish significance

Case studies
Breast cancer DNA methylation (Unsupervised)Breast cancer ATAC-seq (Unsupervised)
Kidney cancer DNA methylation (Supervised)
Pan-cancer EZH2 analysis
Breast cancer DNA methylation PCA

COCOA results for PC1

ER-related regions have higher loadings on PC1
Raw DNA Methylation in ER binding regions

COCOA results for PC1-4

COCOA results for PC1-4

COCOA meta-region plots for PC1-4

Case studies
Breast cancer DNA methylation (Unsupervised)Breast cancer ATAC-seq (Unsupervised)
Kidney cancer DNA methylation (Supervised)
Pan-cancer EZH2 analysis
Breast cancer ATAC-seq PCA

COCOA results for ATAC-seq

ER-related regions have higher loadings on PC1
COCOA results for ATAC-seq

Case studies
Breast cancer DNA methylation (Unsupervised)Breast cancer ATAC-seq (Unsupervised)
Kidney cancer DNA methylation (Supervised)
Pan-cancer EZH2 analysis
Kidney cancer DNA methylation (Supervised)

Rank region sets for methylation
that correlates with cancer stage
COCOA results for cancer stage

COCOA results for cancer stage

COCOA results for cancer stage

Case studies
Breast cancer DNA methylation (Unsupervised)Breast cancer ATAC-seq (Unsupervised)
Kidney cancer DNA methylation (Supervised)
Pan-cancer EZH2 analysis
DNA methylation in EZH2 regions and survival

DNA methylation in EZH2-binding regions most often positively correlated with risk of death.
An optimized ATAC-seq pipeline
with serial alignments

Jason Smith

PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
$ /pipelines/pepatac.py -h
usage: pepatac.py [-h] [-R] [-N] [-D] [-F] [-C CONFIG_FILE]
[-O PARENT_OUTPUT_FOLDER] [-M MEMORY_LIMIT]
[-P NUMBER_OF_CORES] -S SAMPLE_NAME -I INPUT_FILES
[INPUT_FILES ...] [-I2 [INPUT_FILES2 [INPUT_FILES2 ...]]] -G
GENOME_ASSEMBLY [-Q SINGLE_OR_PAIRED] [-gs GENOME_SIZE]
[--frip-ref-peaks FRIP_REF_PEAKS] [--TSS-name TSS_NAME]
[--anno-name ANNO_NAME] [--keep]
[--peak-caller {fseq,macs2}]
[--trimmer {trimmomatic,skewer}]
[--prealignments PREALIGNMENTS [PREALIGNMENTS ...]] [-V]
PEPATAC version 0.7.3
optional arguments:
-h, --help show this help message and exit
-R, --recover Overwrite locks to recover from previous failed run
-N, --new-start Overwrite all results to start a fresh run
-D, --dirty Don't auto-delete intermediate files
-F, --force-follow Always run 'follow' commands
-C CONFIG_FILE, --config CONFIG_FILE
Pipeline configuration file (YAML). Relative paths are
with respect to the pipeline script.
-O PARENT_OUTPUT_FOLDER, --output-parent PARENT_OUTPUT_FOLDER
Parent output directory of project
-M MEMORY_LIMIT, --mem MEMORY_LIMIT
Memory limit (in Mb) for processes accepting such
-P NUMBER_OF_CORES, --cores NUMBER_OF_CORES
Number of cores for parallelized processes
-I2 [INPUT_FILES2 [INPUT_FILES2 ...]], --input2 [INPUT_FILES2 [INPUT_FILES2 ...]]
Secondary input files, such as read2
-Q SINGLE_OR_PAIRED, --single-or-paired SINGLE_OR_PAIRED
Single- or paired-end sequencing protocol
-gs GENOME_SIZE, --genome-size GENOME_SIZE
genome size for MACS2
--frip-ref-peaks FRIP_REF_PEAKS
Reference peak set for calculating FRiP
--TSS-name TSS_NAME Name of TSS annotation file
--anno-name ANNO_NAME
Name of reference bed file for calculating FRiF
--keep Keep prealignment BAM files
--peak-caller {fseq,macs2}
Name of peak caller
--trimmer {trimmomatic,pyadapt,skewer}
Name of read trimming program
--prealignments PREALIGNMENTS [PREALIGNMENTS ...]
Space-delimited list of reference genomes to align to
before primary alignment.
-V, --version show program's version number and exit
required named arguments:
-S SAMPLE_NAME, --sample-name SAMPLE_NAME
Name for sample to run
-I INPUT_FILES [INPUT_FILES ...], --input INPUT_FILES [INPUT_FILES ...]
One or more primary input files
-G GENOME_ASSEMBLY, --genome GENOME_ASSEMBLY
Identifier for genome assembly


PEP specification for sample metadata
1. Configuration file:config.yaml
pep_version: 2.0.0
sample_table: "path/to/sample_table.csv"
sample_table.csv:
"sample_name", "protocol", "file"
"frog_1", "ATAC-seq", "frog1.fq.gz"
"frog_2", "ATAC-seq", "frog2.fq.gz"
"frog_3", "ATAC-seq", "frog3.fq.gz"
"frog_4", "ATAC-seq", "frog4.fq.gz"
MapReduce or Scatter/Gather
1. Map/Scatter PEPATAC across individual sampleslooper run config.yaml
looper runp config.yaml
PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Nuclear-mitochondrial DNA (NuMts) confuse aligners









PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Flexibility and Portability
pepatac.yaml# basic tools
tools: # absolute paths to required tools
java: java
python: python
samtools: samtools
bedtools: bedtools
bowtie2: bowtie2
fastqc: fastqc
macs2: macs2
picard: ${PICARD}
skewer: skewer
perl: perl
# ucsc tools
bedGraphToBigWig: bedGraphToBigWig
wigToBigWig: wigToBigWig
bigWigCat: bigWigCat
bedSort: bedSort
bedToBigBed: bedToBigBed
# optional tools
fseq: fseq
trimmo: ${TRIMMOMATIC}
Rscript: Rscript
# user configure
resources:
genomes: ${GENOMES}
adapters: null # Set to null to use default adapters
parameters: # parameters passed to bioinformatic tools
samtools:
q: 10
macs2:
f: BED
q: 0.01
shift: 0
fseq:
of: npf # narrowPeak as output format
l: 600 # feature length
t: 4.0 # "threshold" (standard deviations)
s: 1 # wiggle track stepPEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Output

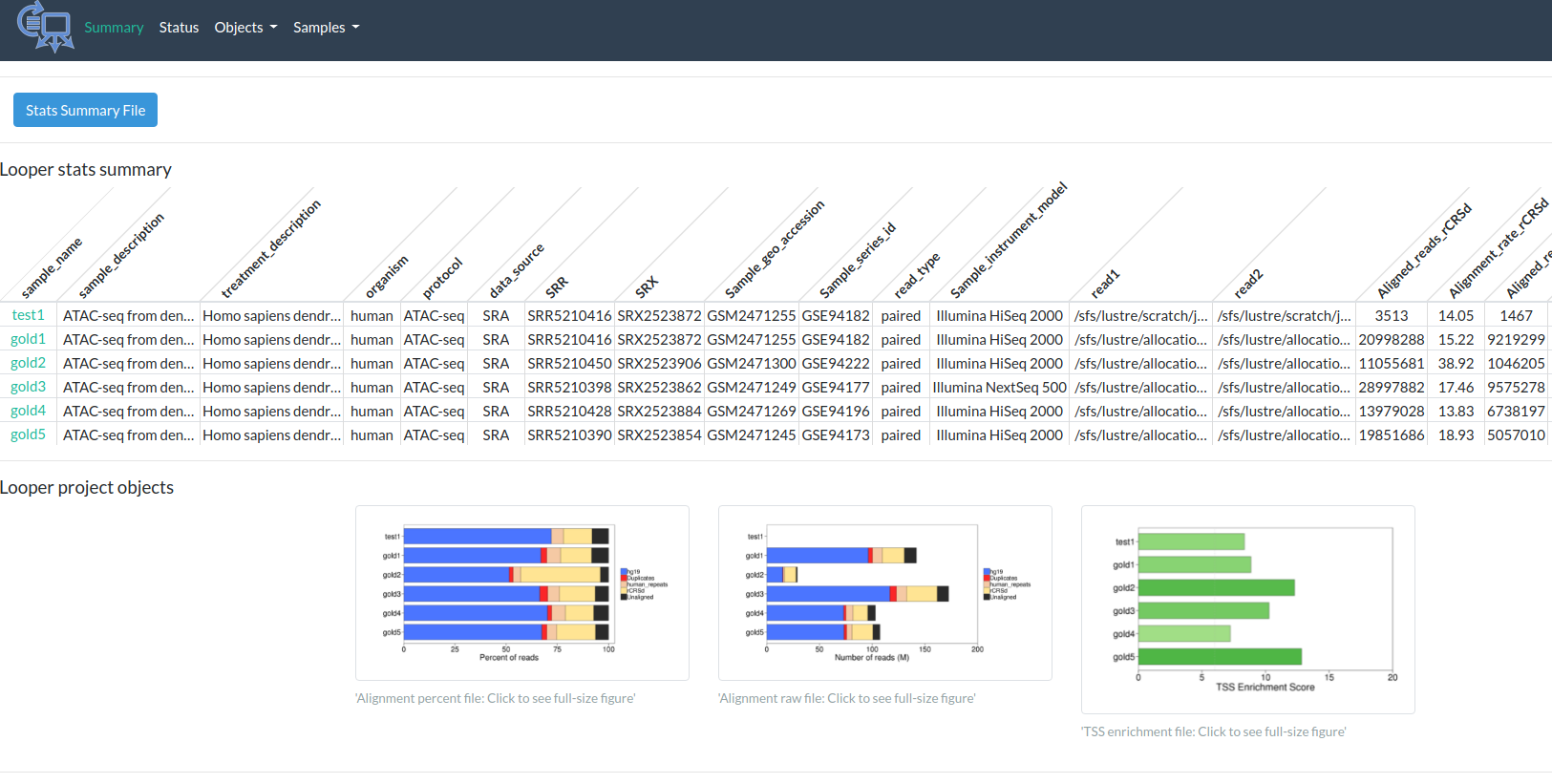
http://pepatac.databio.org/en/latest/files/examples/gold/gold_summary.html
PEPATAC in practice
A full-service reference genome manager.
The problem
Many tools require genome-related assets (like indexes).How should we organize these on disk?

Refgenie consists of 3 components


Refgenie splits tasks between CLI and server

The build/pull method needs provenance checks
Asset provenance:

Genome provenance:

Refget
Refget enables access to reference sequencesusing an identifier derived from the sequence itself.
How refget works

Refget v2.0: Collections for genome provenance

Recursive checksums have advantages
Allows getting content list only
Preserves chromosome order
Re-uses the checksum function
Duplicates are stored only once
Go one step further for...
Preserves chromosome order
Re-uses the checksum function
Duplicates are stored only once
Go one step further for...

It keeps going... and going...

Asset provenance:
Recipes + containers?
Genome provenance:
Solved by refget v2.0?
Tying human identifiers to a digest:
hg38:
refget_digest: 32a37a52a377d95bfd4b3d66763e1396a4480f34ab5c318a
Pepkit
A structure and toolkit for organizing large-scale,sample-intensive biological research projects
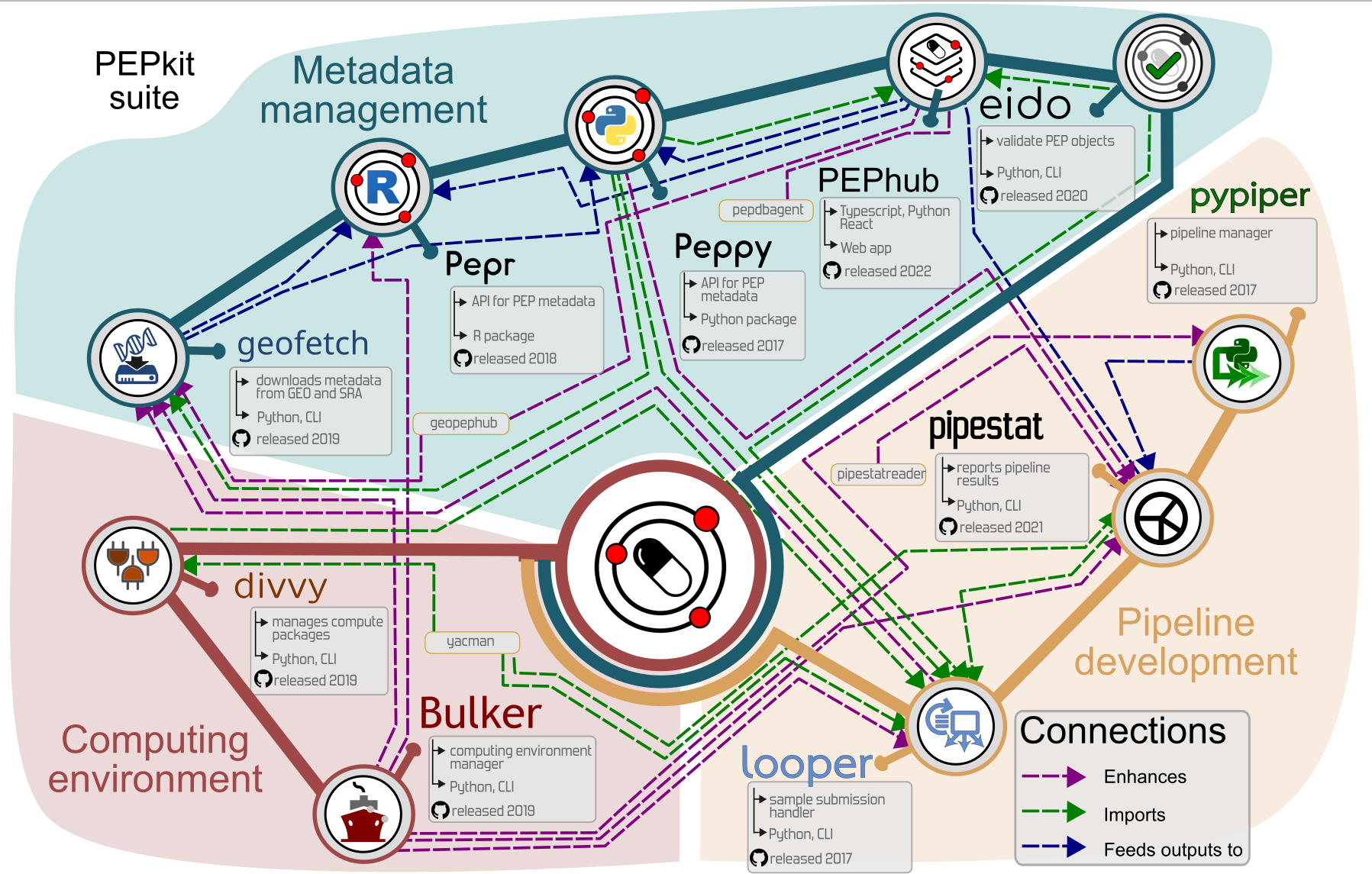
Research is organized in projects


How do we conceptualize a research project?
Each project has 3 components

Organizing multiple projects is a challenge

How do I re-use a component?

A project is a set of edges in a tripartite graph

Enable linking with interfaces

We are building a modular ecosystem

pepkit · geofetch · looper · caravel · pypiper · divvy
PEP: Portable Encapsulated Projects
 PEP format
PEP format
sample_name,protocol,organism,input_file
frog_0h,RNA-seq,frog,/path/to/frog0.gz
frog_1h,RNA-seq,frog,/path/to/frog1.gz
frog_2h,RNA-seq,frog,/path/to/frog2.gz
frog_3h,RNA-seq,frog,/path/to/frog3.gz
PEP format
sample_name,protocol,organism,input_file
frog_0h,RNA-seq,frog,/path/to/frog0.gz
frog_1h,RNA-seq,frog,/path/to/frog1.gz
frog_2h,RNA-seq,frog,/path/to/frog2.gz
frog_3h,RNA-seq,frog,/path/to/frog3.gz
sample_table: /path/to/samples.csv
output_dir: /path/to/output/folder
other_variable: value
Add programmatic sample and project modifiers.
Automatically build new sample attributes from existing attributes.
Without derived attribute:
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | /path/to/frog0.gz |
| frog_1h | 1 | RNA-seq | frog | /path/to/frog1.gz |
| frog_2h | 2 | RNA-seq | frog | /path/to/frog2.gz |
| frog_3h | 3 | RNA-seq | frog | /path/to/frog3.gz |
Using derived attribute:
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | my_samples |
| frog_1h | 1 | RNA-seq | frog | my_samples |
| frog_2h | 2 | RNA-seq | frog | my_samples |
| frog_3h | 3 | RNA-seq | frog | my_samples |
| crab_0h | 0 | RNA-seq | crab | your_samples |
| crab_3h | 3 | RNA-seq | crab | your_samples |
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | my_samples |
| frog_1h | 1 | RNA-seq | frog | my_samples |
| frog_2h | 2 | RNA-seq | frog | my_samples |
| frog_3h | 3 | RNA-seq | frog | my_samples |
| crab_0h | 0 | RNA-seq | crab | your_samples |
| crab_3h | 3 | RNA-seq | crab | your_samples |
Project config file:
sample_modifiers:
derive:
attributes: [input_file]
sources:
my_samples: "/path/to/my/samples/{organism}_{t}h.gz"
your_samples: "/path/to/your/samples/{organism}_{t}h.gz"Benefit: Enables distributed files, portability
Add new sample attributes conditioned on values of existing attributes
Before:
| sample_name | protocol | organism |
| ------------- | :-------------: | -------- |
| human_1 | RNA-seq | human |
| human_2 | RNA-seq | human |
| human_3 | RNA-seq | human |
| mouse_1 | RNA-seq | mouse |
After:
| sample_name | protocol | organism | genome |
| ------------- | :-------------: | -------- | ------ |
| human_1 | RNA-seq | human | hg38 |
| human_2 | RNA-seq | human | hg38 |
| human_3 | RNA-seq | human | hg38 |
| mouse_1 | RNA-seq | mouse | mm10 |
| sample_name | protocol | organism |
| ------------- | :-------------: | -------- |
| human_1 | RNA-seq | human |
| human_2 | RNA-seq | human |
| human_3 | RNA-seq | human |
| mouse_1 | RNA-seq | mouse |
Project config file:
sample_modifiers:
imply:
- if:
organism: human
then:
genome: hg38
- if:
organism: mouse
then:
genome: mm10Benefit: Divides project from sample metadata
Define activatable project attributes.
project_modifiers:
amendments:
diverse:
metadata:
sample_annotation: psa_rrbs_diverse.csv
cancer:
metadata:
sample_annotation: psa_rrbs_intracancer.csvBenefit: Defines multiple similar projects in a single file
Reproducibility
data + code+ environment
Containers
A promising solution, but how should we use them?
Combined

Individual

| easy to deploy |
| easy to use |
| reusable |
| combinable |
| subsetable |
| space efficient |
| Combined |
| Individual |
| Bulker |
How bulker does it
Two conceptual advances:
Containerized executables

Distribute containers in sets

Thank You
Collaborators
Vince Reuter
Andre Rendeiro
Levi Waldron
Alumni
Aaron Gu
Jianglin Feng
Ognen Duzlevski
Tessa Danehy
Vince Reuter
Andre Rendeiro
Levi Waldron
Alumni
Aaron Gu
Jianglin Feng
Ognen Duzlevski
Tessa Danehy
Sheffield lab
Erfaneh Gharavi
Michal Stolarczyk
John Lawson
Jason Smith
Kristyna Kupkova
John Stubbs
Bingjie Xue
Jose Verdezoto
Nathan LeRoy
Oleksandr Khoroshevskyi
Erfaneh Gharavi
Michal Stolarczyk
John Lawson
Jason Smith
Kristyna Kupkova
John Stubbs
Bingjie Xue
Jose Verdezoto
Nathan LeRoy
Oleksandr Khoroshevskyi
Funding:



NIGMS R35-GM128636
NIGMS R35-GM128636