Downstream ATAC-seq analysis
Nathan Sheffield, PhDCommon types of downstream ATAC analysis
- Determining differential chromatin accessibility
- Clustering elements that share accessibility patterns
- Footprinting
- Region of Interest investigation
Differential ATAC-seq analysis

How to create the count matrix? 1) Consensus peaks; 2) tiles.
Consensus peaks

Genome tiles

Hybrid: Smart tiles

Statistical tests
Use count-based statistical tools for RNA-seq: DEseq or edgeR.
Clustering ATAC-seq data

Chromatin accessibility across cell types
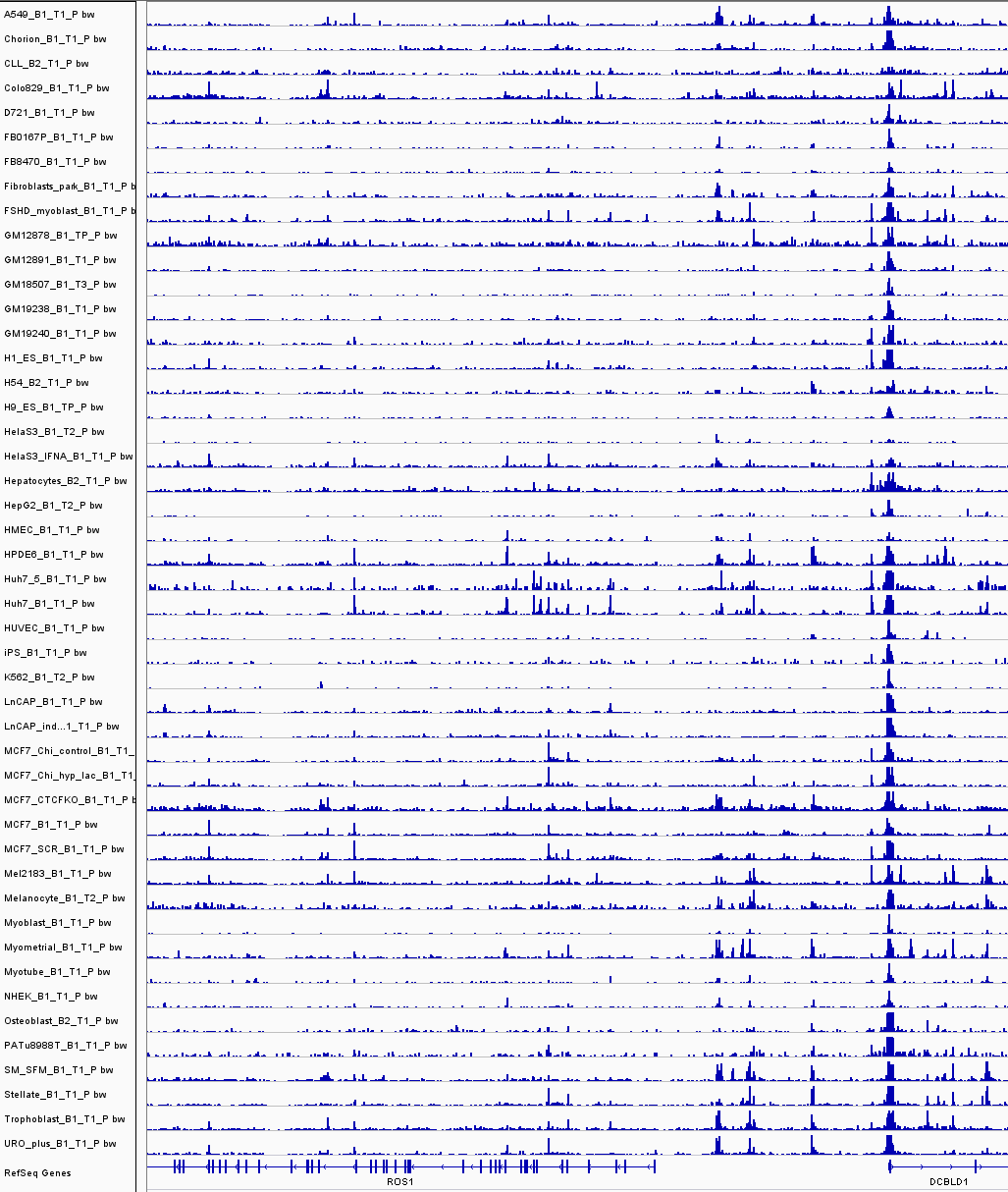
These still require 'common reference peaks'...
(Ibrahim et al. 2015)
(Tarbell and Liu 2019)
It is possible to extend HMMRATAC to identify differentially accessible regions between two or more conditions... (Tarbell and Liu 2019)
Footprinting
Footprinting concept
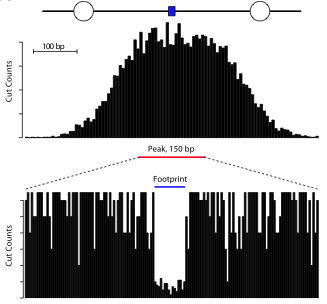
Vernon et al. 2012
Caveats: 1) Depth; 2) Sequence bias; 3) Factor dynamics
Bias correction
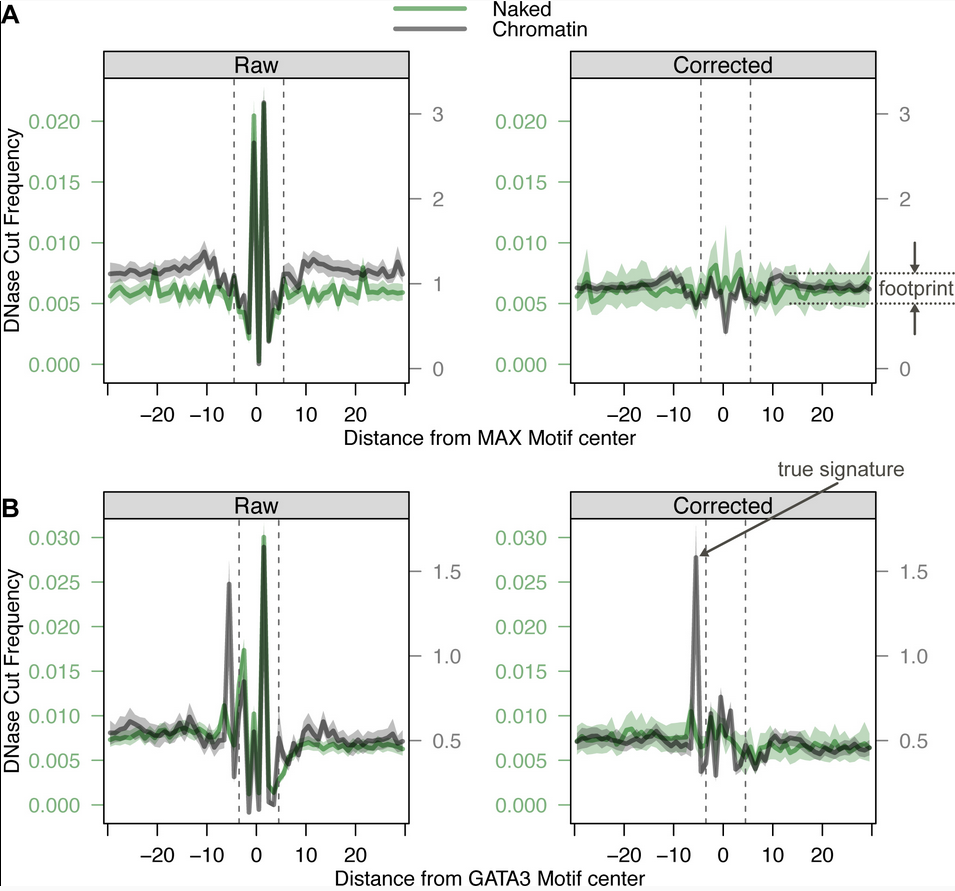
Martins et al. 2018
Factor dynamics
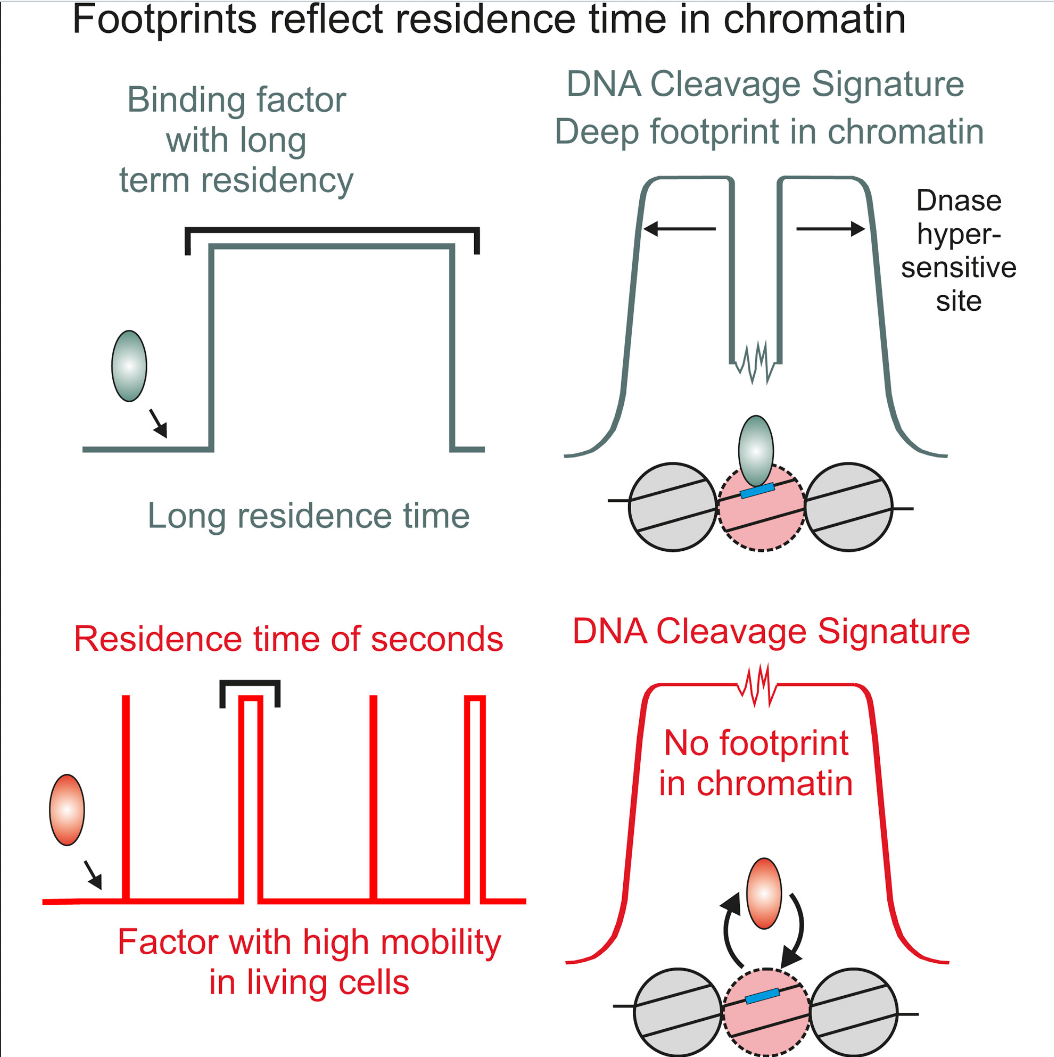
Sung et al. 2014
Single-cell ATAC
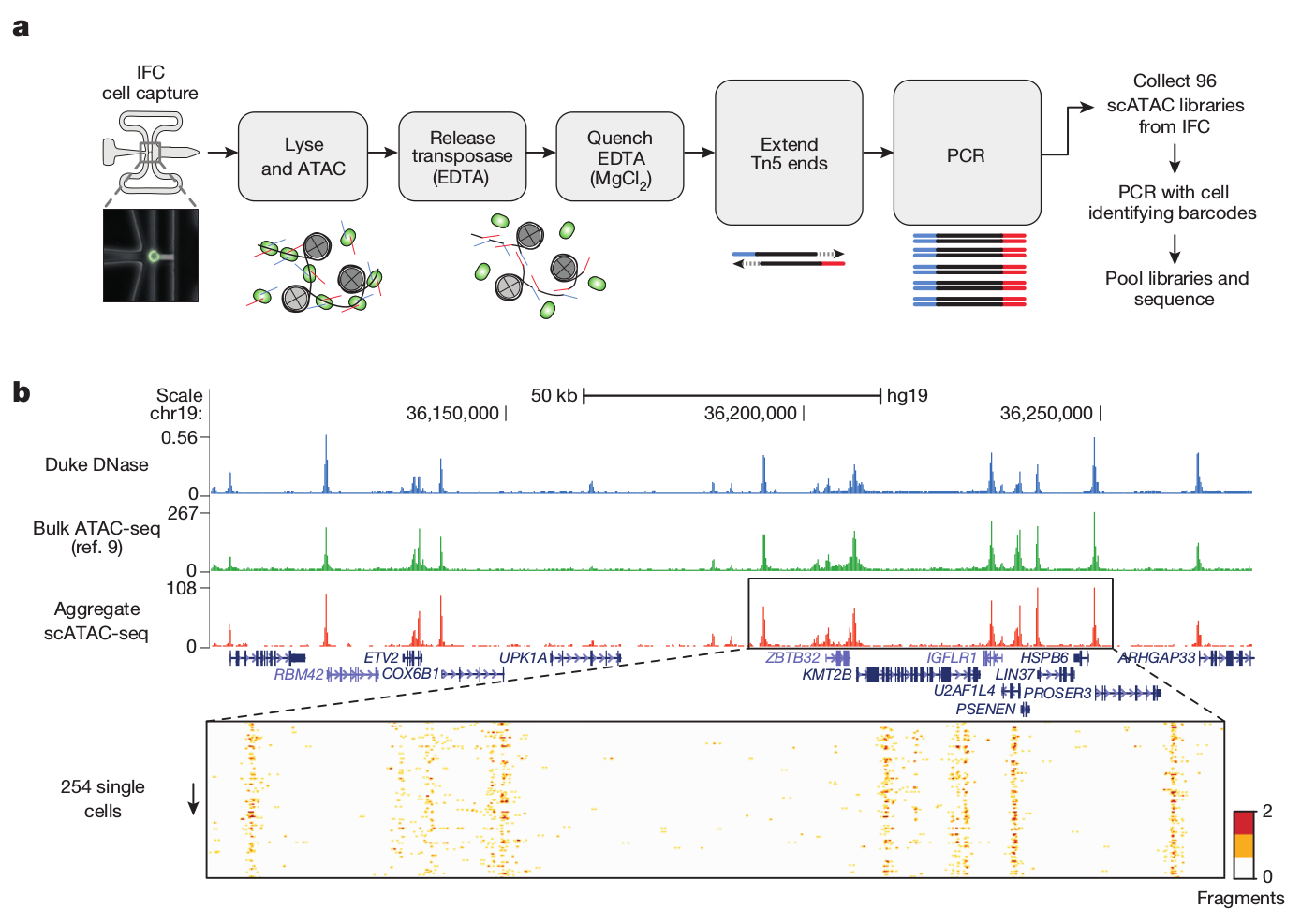
Buenrostro et al. 2015
Xiong et al. 2019 - SCALE method for single-cell ATAC-seq analysis via latent feature extraction




