New computational tools for epigenome analysis
Nathan Sheffield, PhDOutline
PEPATAC
Regionset2vec
|
|
40%
40%
20%
|
GA4GH Sequence Collections
◁ Questions ▷
An optimized ATAC-seq pipeline
with serial alignments

Jason Smith

PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
$ /pipelines/pepatac.py -h
usage: pepatac.py [-h] [-R] [-N] [-D] [-F] [-C CONFIG_FILE]
[-O PARENT_OUTPUT_FOLDER] [-M MEMORY_LIMIT]
[-P NUMBER_OF_CORES] -S SAMPLE_NAME -I INPUT_FILES
[INPUT_FILES ...] [-I2 [INPUT_FILES2 [INPUT_FILES2 ...]]] -G
GENOME_ASSEMBLY [-Q SINGLE_OR_PAIRED] [-gs GENOME_SIZE]
[--frip-ref-peaks FRIP_REF_PEAKS] [--TSS-name TSS_NAME]
[--anno-name ANNO_NAME] [--keep]
[--peak-caller {fseq,macs2}]
[--trimmer {trimmomatic,skewer}]
[--prealignments PREALIGNMENTS [PREALIGNMENTS ...]] [-V]
PEPATAC version 0.7.3
optional arguments:
-h, --help show this help message and exit
-R, --recover Overwrite locks to recover from previous failed run
-N, --new-start Overwrite all results to start a fresh run
-D, --dirty Don't auto-delete intermediate files
-F, --force-follow Always run 'follow' commands
-C CONFIG_FILE, --config CONFIG_FILE
Pipeline configuration file (YAML). Relative paths are
with respect to the pipeline script.
-O PARENT_OUTPUT_FOLDER, --output-parent PARENT_OUTPUT_FOLDER
Parent output directory of project
-M MEMORY_LIMIT, --mem MEMORY_LIMIT
Memory limit (in Mb) for processes accepting such
-P NUMBER_OF_CORES, --cores NUMBER_OF_CORES
Number of cores for parallelized processes
-I2 [INPUT_FILES2 [INPUT_FILES2 ...]], --input2 [INPUT_FILES2 [INPUT_FILES2 ...]]
Secondary input files, such as read2
-Q SINGLE_OR_PAIRED, --single-or-paired SINGLE_OR_PAIRED
Single- or paired-end sequencing protocol
-gs GENOME_SIZE, --genome-size GENOME_SIZE
genome size for MACS2
--frip-ref-peaks FRIP_REF_PEAKS
Reference peak set for calculating FRiP
--TSS-name TSS_NAME Name of TSS annotation file
--anno-name ANNO_NAME
Name of reference bed file for calculating FRiF
--keep Keep prealignment BAM files
--peak-caller {fseq,macs2}
Name of peak caller
--trimmer {trimmomatic,pyadapt,skewer}
Name of read trimming program
--prealignments PREALIGNMENTS [PREALIGNMENTS ...]
Space-delimited list of reference genomes to align to
before primary alignment.
-V, --version show program's version number and exit
required named arguments:
-S SAMPLE_NAME, --sample-name SAMPLE_NAME
Name for sample to run
-I INPUT_FILES [INPUT_FILES ...], --input INPUT_FILES [INPUT_FILES ...]
One or more primary input files
-G GENOME_ASSEMBLY, --genome GENOME_ASSEMBLY
Identifier for genome assembly


PEP specification for sample metadata
1. Configuration file:config.yaml
pep_version: 2.0.0
sample_table: "path/to/sample_table.csv"
sample_table.csv:
"sample_name", "protocol", "file"
"frog_1", "ATAC-seq", "frog1.fq.gz"
"frog_2", "ATAC-seq", "frog2.fq.gz"
"frog_3", "ATAC-seq", "frog3.fq.gz"
"frog_4", "ATAC-seq", "frog4.fq.gz"
MapReduce or Scatter/Gather
1. Map/Scatter PEPATAC across individual sampleslooper run config.yaml
looper runp config.yaml
PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Nuclear-mitochondrial DNA (NuMts) confuse aligners









PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Flexibility and Portability
pepatac.yaml# basic tools
tools: # absolute paths to required tools
java: java
python: python
samtools: samtools
bedtools: bedtools
bowtie2: bowtie2
fastqc: fastqc
macs2: macs2
picard: ${PICARD}
skewer: skewer
perl: perl
# ucsc tools
bedGraphToBigWig: bedGraphToBigWig
wigToBigWig: wigToBigWig
bigWigCat: bigWigCat
bedSort: bedSort
bedToBigBed: bedToBigBed
# optional tools
fseq: fseq
trimmo: ${TRIMMOMATIC}
Rscript: Rscript
# user configure
resources:
genomes: ${GENOMES}
adapters: null # Set to null to use default adapters
parameters: # parameters passed to bioinformatic tools
samtools:
q: 10
macs2:
f: BED
q: 0.01
shift: 0
fseq:
of: npf # narrowPeak as output format
l: 600 # feature length
t: 4.0 # "threshold" (standard deviations)
s: 1 # wiggle track stepPEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Output

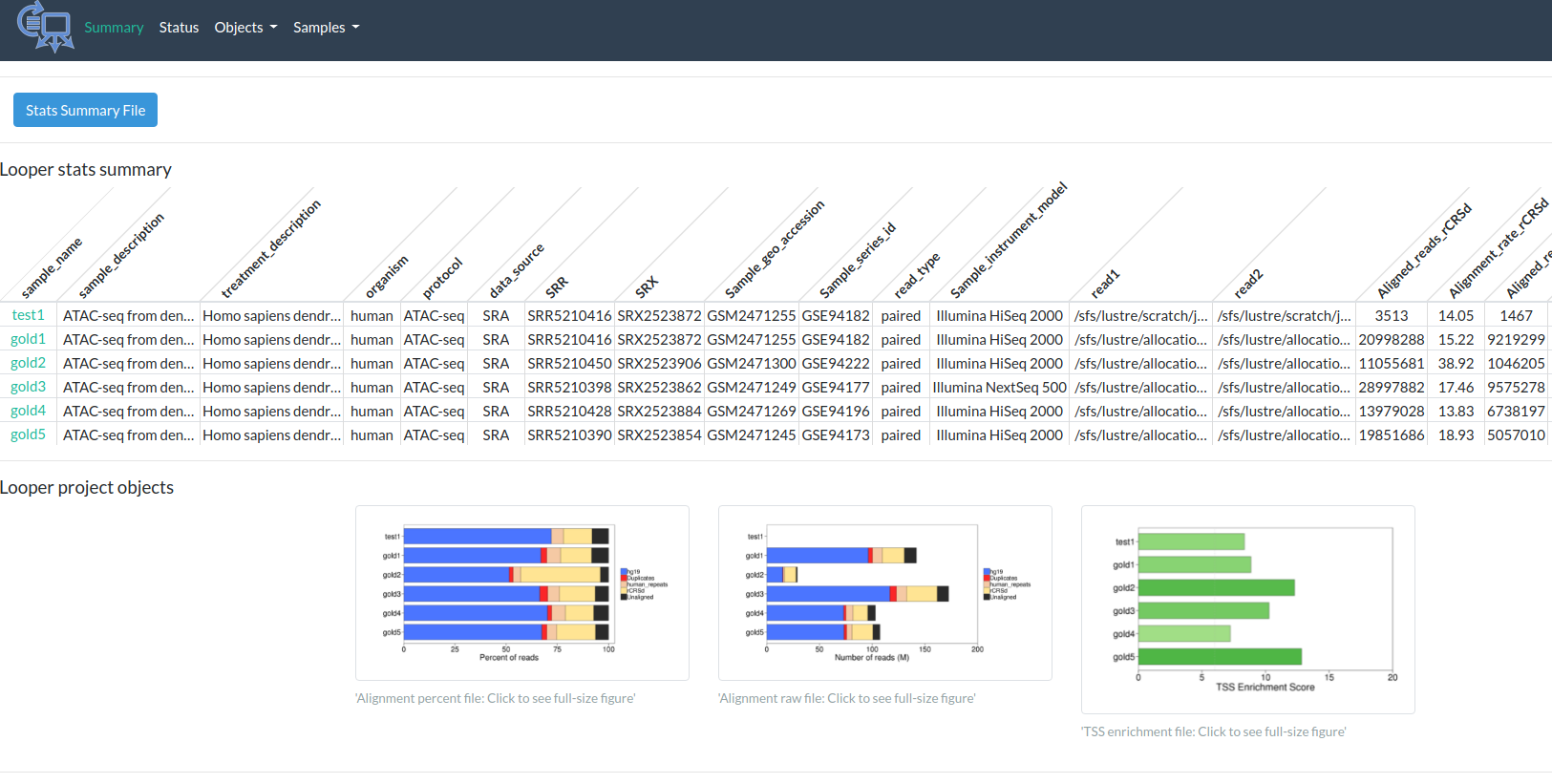
http://pepatac.databio.org/en/latest/files/examples/gold/gold_summary.html
PEPATAC in practice
Unique identifiers and API for sequence collections.
Problem
Who is the authoritative provider of the reference genome?
- NCBI?
- UCSC?
- Ensembl?
- hard, soft, or no repeat masking?
- are alternative scaffolds included?
- are haplotypes included?
- how are chromosomes named (chr1, 1, or NC_000001.11)?
- how is the assembly named (hg38, GRCh38, or GCF_000001405.39)?
- Are any decoy sequences included (like EBV)?
| Provider | Chr1 name | Chr1 length | Chr1 md5 | Num chroms |
|---|---|---|---|---|
| Ensembl primary | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 195 |
| Ensembl toplevel | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 649 |
| NCBI | NC_000001.11 | 248956422 | 6aef897c3d6ff0c78aff06ac189178dd | 640 |
| UCSC | chr1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 456 |
https://gist.github.com/andrewyatz/692f81baab1bebaf09c481937f2ad6c6
Subtle differences in reference assembly lead to:
- Lack of reproducibility of analysis
- Lack of reusability of results
Solution

Refget -> Sequence collections
Refget
Refget enables access to reference sequencesusing an identifier derived from the sequence itself.
How refget works

Limitations
- only handles a single sequence
- excludes chromosome names
- no capacity for annotation
Extending to sequence collections
We need:- 1. An algorithm to create a deterministic, unique digest from a collection of sequences
- 2. A server capable of retrieving sequences given an identifier
First pass: Refgenie approach


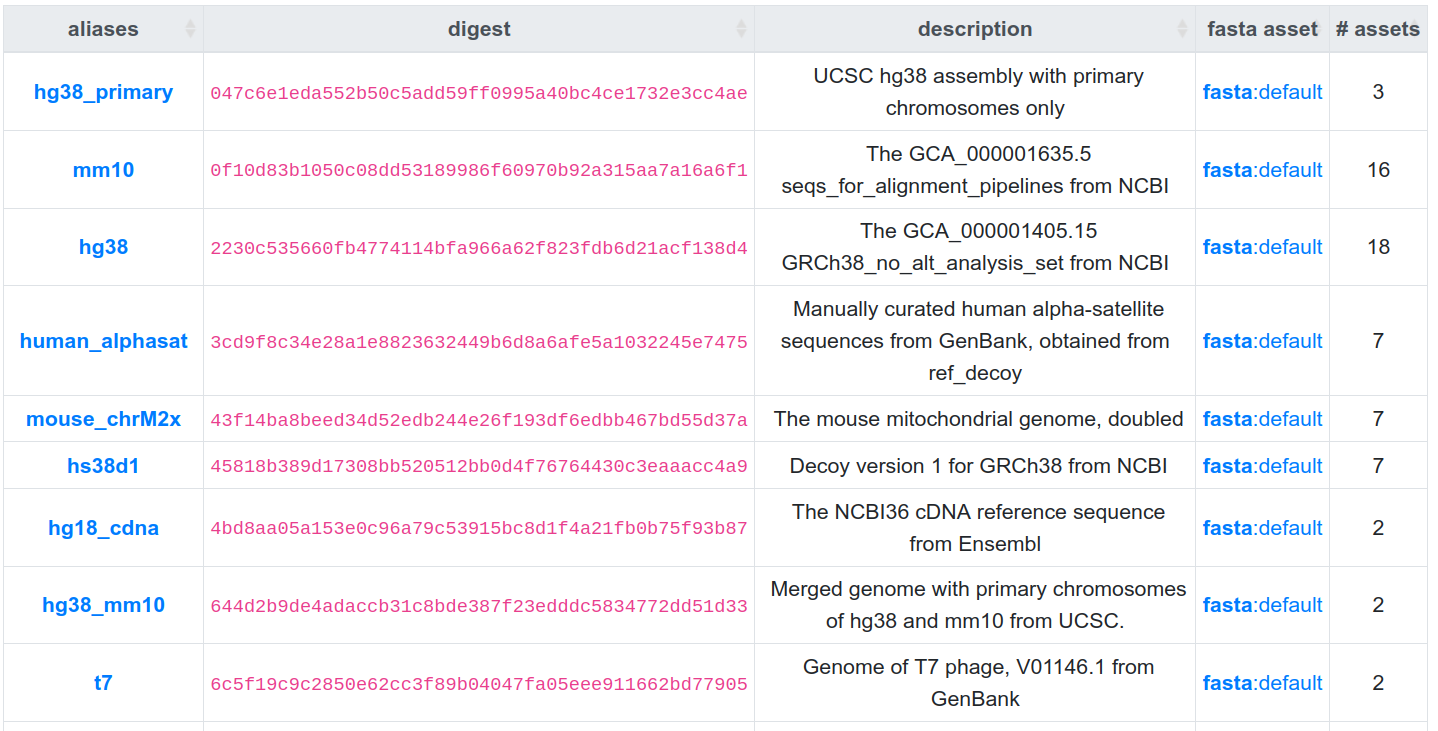
Limitations and discussion
- Should we include sequence topology in the digest?
- What other attributes could we include?
- Are there better delimiters?
- How do we construct the 'string-to-digest'?
- How do we handle order of sequences?
- How should the API respond to requests?
Project goal:
The project specifies:
How do we digest a sequence collection?
JSON object: each sequence collection attribute is a property
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
],
"sequences": [
"31fc6ca291a32fb9df82b85e5f077e31",
"92c6a56c9e9459d8a42b96f7884710bc",
"5f63cfaa3ef61f88c9635fb9d18ec945"
]
}
← length of the sequences
← names of the sequences
← refget digests
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
],
"sequences": [
"31fc6ca291a32fb9df...",
"92c6a56c9e9459d8a4...",
"5f63cfaa3ef61f88c9..."
]
}
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
]
}
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
],
"sequences": [
"31fc6ca291a32fb9df...",
"92c6a56c9e9459d8a4...",
"5f63cfaa3ef61f88c9..."
],
"topologies" [
"linear",
"linear",
"circular"
]
}
Digest algorithm
- Canonicalize each attribute following RFC-8785 (JSON Canonicalization Scheme)
- Digest each string (GA4GH digest: SHA512 truncated to 24 bits, converted to base64)
- Canonicalize the entire object
- Digest the canonicalized string
 Tim Cezard
Tim Cezard
Advantages
- Accommodates new attributes with backwards-compatibility
- Additional layer of recursion to assess individual attributes
- Relies on existing JCS standard for string encoding
What gets digested?
Comparison function
| Provider | Chr1 name | Chr1 length | Chr1 md5 | Num chroms |
|---|---|---|---|---|
| Ensembl primary | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 195 |
| Ensembl toplevel | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 649 |
| NCBI | NC_000001.11 | 248956422 | 6aef897c3d6ff0c78aff06ac189178dd | 640 |
| UCSC | chr1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 456 |
- seqcol 1: 047c6e1eda552b50c5add59ff0995
- seqcol 2: 2230c535660fb4774114bfa966a62
How compatible are they?
Comparison endpoint
{
"digests": {
"a": "59319772d1bcf2e0dd4b8a296f2d9682",
"b": "2e7bc302a54ecec62d8155e19fbf2748"
},
"arrays": {
"a-only": [],
"b-only": [],
"a-and-b": [
"lengths",
"names",
"sequences",
"names_lengths"
]
},
"elements": {
"total": {
"a": 3,
"b": 3
},
"a-and-b": {
"lengths": 3,
"names": 3,
"sequences": 3,
"names_lengths": 3
},
"a-and-b-same-order": {
"lengths": false,
"names": false,
"sequences": false,
"names_lengths": true
}
}
}
Seqcol API demonstration
https://seqcolapi.databio.org/API endpoints
GET /service-infoGET /collection/:digestGET /comparison/:digest1/:digest2POST /comparison/:digest1Conclusions
- Refget provides universal IDs for individual sequences
- Sequence collections extends this to reference genomes
- Using a deterministic algorithm, you can find the identifier
- A lookup service can retrieve the original sequence
- A comparison function allows fine-grained compatibility tests
- Please follow along: https://github.com/ga4gh/seqcol-spec
Region-set 2 Vec
Embeddings of genomic region setsin lower dimensions.

Erfaneh Gharavi
Word embeddings
http://suriyadeepan.github.io
Word2vec model
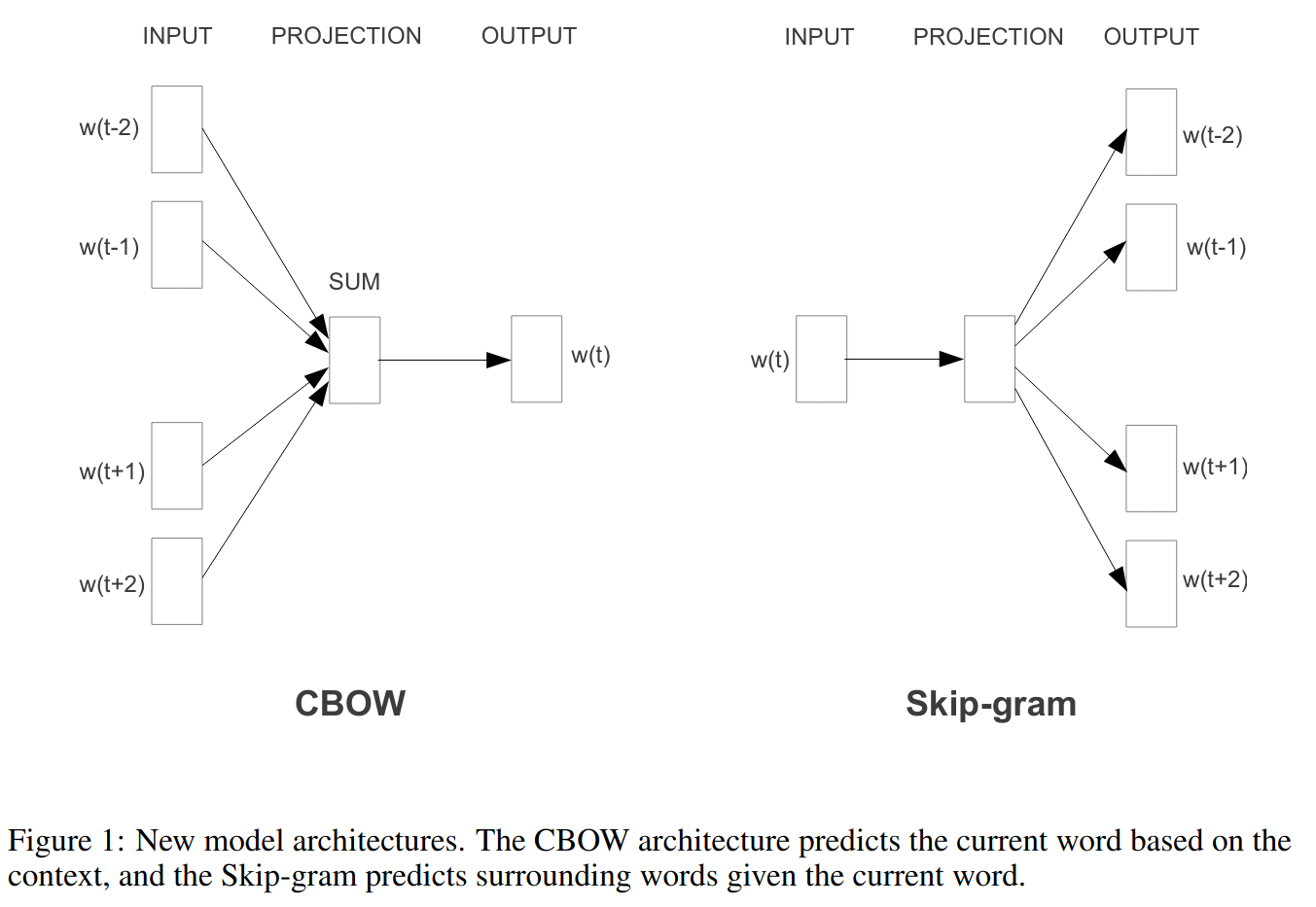
Word context
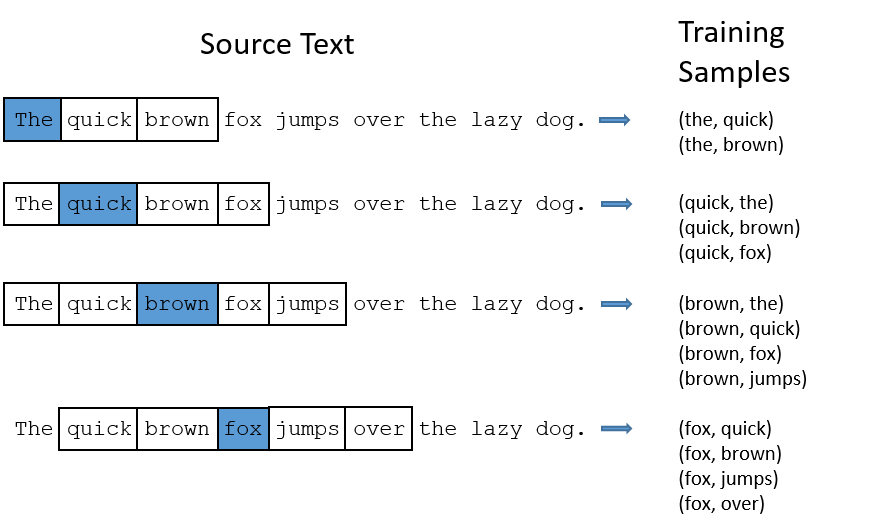
You shall know a word by the company it keeps. (Firth 1957)
Words that occur in similar contexts tend to have similar meanings.
Words that occur in similar contexts tend to have similar meanings.
Image credit: Shubham Agarwal
Genomic context
A genomic interval is more likely to appear in a BED file with other genomic intervals of a similar function.

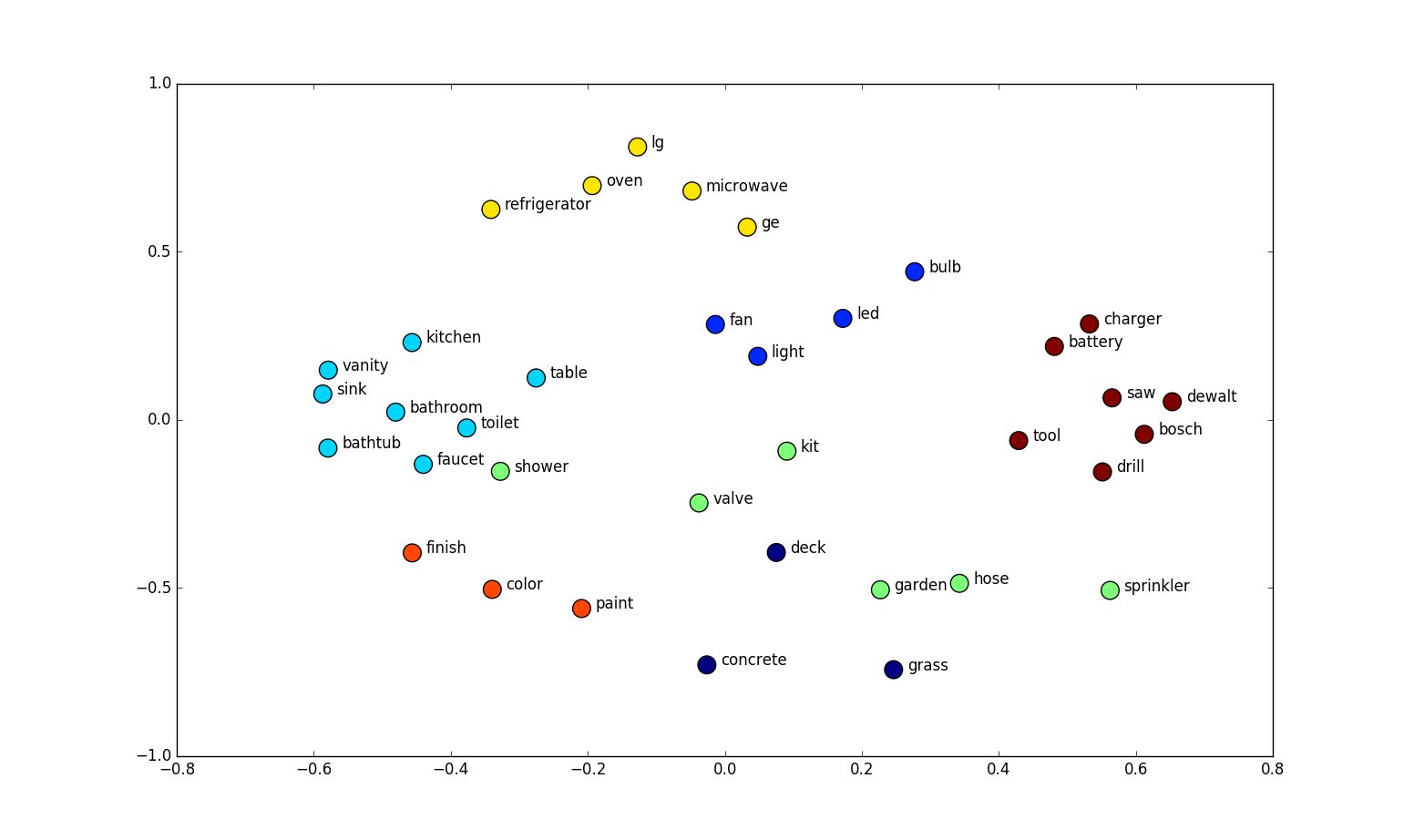
Genomic Interval Embeddings

Evaluation
We have created unsupervised 100-dimensional vector representations (embeddings) of region sets.Do relationships among vectors reflect biology?

Evaluation 1: Classification performance

Evaluation 1: Classification performance

Evaluation 1: Classification performance


Conclusion
- Regionset2vec adapts word2vec to learn genomic region embeddings
- Regionset2vec embeddings capture biological information
- NLP approaches can be adapted for applications in genomic interval analysis
Next steps
Region embeddings help with:Given a BED file, find a similar BED file.
(LOLA, GIGGLE, IGD)
What about:
Given a human search term, find a BED file.
Given a human search term, find a BED file.
Thank You
Collaborators
Aakrosh Ratan
Aidong Zhang
Guangtao Zheng
Don Brown
Hyun Jae Cho
Vince Carey
Mikhail Dozmorov
GA4GH Refget group
Alumni
Aaron Gu
Jianglin Feng
Ognen Duzlevski
Tessa Danehy
Aakrosh Ratan
Aidong Zhang
Guangtao Zheng
Don Brown
Hyun Jae Cho
Vince Carey
Mikhail Dozmorov
GA4GH Refget group
Alumni
Aaron Gu
Jianglin Feng
Ognen Duzlevski
Tessa Danehy
Sheffield lab
Erfaneh Gharavi
Michal Stolarczyk
John Lawson
Jason Smith
Kristyna Kupkova
John Stubbs
Bingjie Xue
Jose Verdezoto
Nathan LeRoy
Oleksandr Khoroshevskyi
Erfaneh Gharavi
Michal Stolarczyk
John Lawson
Jason Smith
Kristyna Kupkova
John Stubbs
Bingjie Xue
Jose Verdezoto
Nathan LeRoy
Oleksandr Khoroshevskyi
Funding:



NIGMS R35-GM128636
NIGMS R35-GM128636