ATAC-seq analysis via pipeline
Nathan Sheffield, PhDOutline
Pipelines
ATAC-seq QC
|
|
20%
50%
30%
|
ATAC-seq pipeline
◁ Questions ▷
Analysis spectrum

Advantages: interactive analysis vs pipelines
Interactive
- More universal learning curve
- Direct control
- Quicker to get started
- Easier for simple analysis
- More universal learning curve
- Direct control
- Quicker to get started
- Easier for simple analysis
Pipeline
- Easier for high volume
- More robust error handling
- Automatic logging
- Restartable
- Built-in monitoring
- More repeatable
- More reproducible
- Easier for high volume
- More robust error handling
- Automatic logging
- Restartable
- Built-in monitoring
- More repeatable
- More reproducible
What should I use?
The combination that best fits your project requirements.
ATAC-seq pipelines
There is growing need for integrated pipelines to process ATAC-seq data. Several have been developed but have different focus for downstream analysis by stitching together previously discussed tools. (Yan et al. 2020)

PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
$ /pipelines/pepatac.py -h
usage: pepatac.py [-h] [-R] [-N] [-D] [-F] [-C CONFIG_FILE]
[-O PARENT_OUTPUT_FOLDER] [-M MEMORY_LIMIT]
[-P NUMBER_OF_CORES] -S SAMPLE_NAME -I INPUT_FILES
[INPUT_FILES ...] [-I2 [INPUT_FILES2 [INPUT_FILES2 ...]]] -G
GENOME_ASSEMBLY [-Q SINGLE_OR_PAIRED] [-gs GENOME_SIZE]
[--frip-ref-peaks FRIP_REF_PEAKS] [--TSS-name TSS_NAME]
[--anno-name ANNO_NAME] [--keep]
[--peak-caller {fseq,macs2}]
[--trimmer {trimmomatic,skewer}]
[--prealignments PREALIGNMENTS [PREALIGNMENTS ...]] [-V]
PEPATAC version 0.7.3
optional arguments:
-h, --help show this help message and exit
-R, --recover Overwrite locks to recover from previous failed run
-N, --new-start Overwrite all results to start a fresh run
-D, --dirty Don't auto-delete intermediate files
-F, --force-follow Always run 'follow' commands
-C CONFIG_FILE, --config CONFIG_FILE
Pipeline configuration file (YAML). Relative paths are
with respect to the pipeline script.
-O PARENT_OUTPUT_FOLDER, --output-parent PARENT_OUTPUT_FOLDER
Parent output directory of project
-M MEMORY_LIMIT, --mem MEMORY_LIMIT
Memory limit (in Mb) for processes accepting such
-P NUMBER_OF_CORES, --cores NUMBER_OF_CORES
Number of cores for parallelized processes
-I2 [INPUT_FILES2 [INPUT_FILES2 ...]], --input2 [INPUT_FILES2 [INPUT_FILES2 ...]]
Secondary input files, such as read2
-Q SINGLE_OR_PAIRED, --single-or-paired SINGLE_OR_PAIRED
Single- or paired-end sequencing protocol
-gs GENOME_SIZE, --genome-size GENOME_SIZE
genome size for MACS2
--frip-ref-peaks FRIP_REF_PEAKS
Reference peak set for calculating FRiP
--TSS-name TSS_NAME Name of TSS annotation file
--anno-name ANNO_NAME
Name of reference bed file for calculating FRiF
--keep Keep prealignment BAM files
--peak-caller {fseq,macs2}
Name of peak caller
--trimmer {trimmomatic,pyadapt,skewer}
Name of read trimming program
--prealignments PREALIGNMENTS [PREALIGNMENTS ...]
Space-delimited list of reference genomes to align to
before primary alignment.
-V, --version show program's version number and exit
required named arguments:
-S SAMPLE_NAME, --sample-name SAMPLE_NAME
Name for sample to run
-I INPUT_FILES [INPUT_FILES ...], --input INPUT_FILES [INPUT_FILES ...]
One or more primary input files
-G GENOME_ASSEMBLY, --genome GENOME_ASSEMBLY
Identifier for genome assembly


PEP specification for sample metadata
1. Configuration file:config.yaml
pep_version: 2.0.0
sample_table: "path/to/sample_table.csv"
sample_table.csv:
"sample_name", "protocol", "file"
"frog_1", "ATAC-seq", "frog1.fq.gz"
"frog_2", "ATAC-seq", "frog2.fq.gz"
"frog_3", "ATAC-seq", "frog3.fq.gz"
"frog_4", "ATAC-seq", "frog4.fq.gz"
MapReduce or Scatter/Gather
1. Map/Scatter PEPATAC across individual sampleslooper run config.yaml
looper runp config.yaml
PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Nuclear-mitochondrial DNA (NuMts) confuse aligners









PEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Flexibility and Portability
pepatac.yaml# basic tools
tools: # absolute paths to required tools
java: java
python: python
samtools: samtools
bedtools: bedtools
bowtie2: bowtie2
fastqc: fastqc
macs2: macs2
picard: ${PICARD}
skewer: skewer
perl: perl
# ucsc tools
bedGraphToBigWig: bedGraphToBigWig
wigToBigWig: wigToBigWig
bigWigCat: bigWigCat
bedSort: bedSort
bedToBigBed: bedToBigBed
# optional tools
fseq: fseq
trimmo: ${TRIMMOMATIC}
Rscript: Rscript
# user configure
resources:
genomes: ${GENOMES}
adapters: null # Set to null to use default adapters
parameters: # parameters passed to bioinformatic tools
samtools:
q: 10
macs2:
f: BED
q: 0.01
shift: 0
fseq:
of: npf # narrowPeak as output format
l: 600 # feature length
t: 4.0 # "threshold" (standard deviations)
s: 1 # wiggle track stepPEPATAC strengths
Modular system
Prealignments
Prealignments
Flexibility and portability
Outputs
Outputs
Output

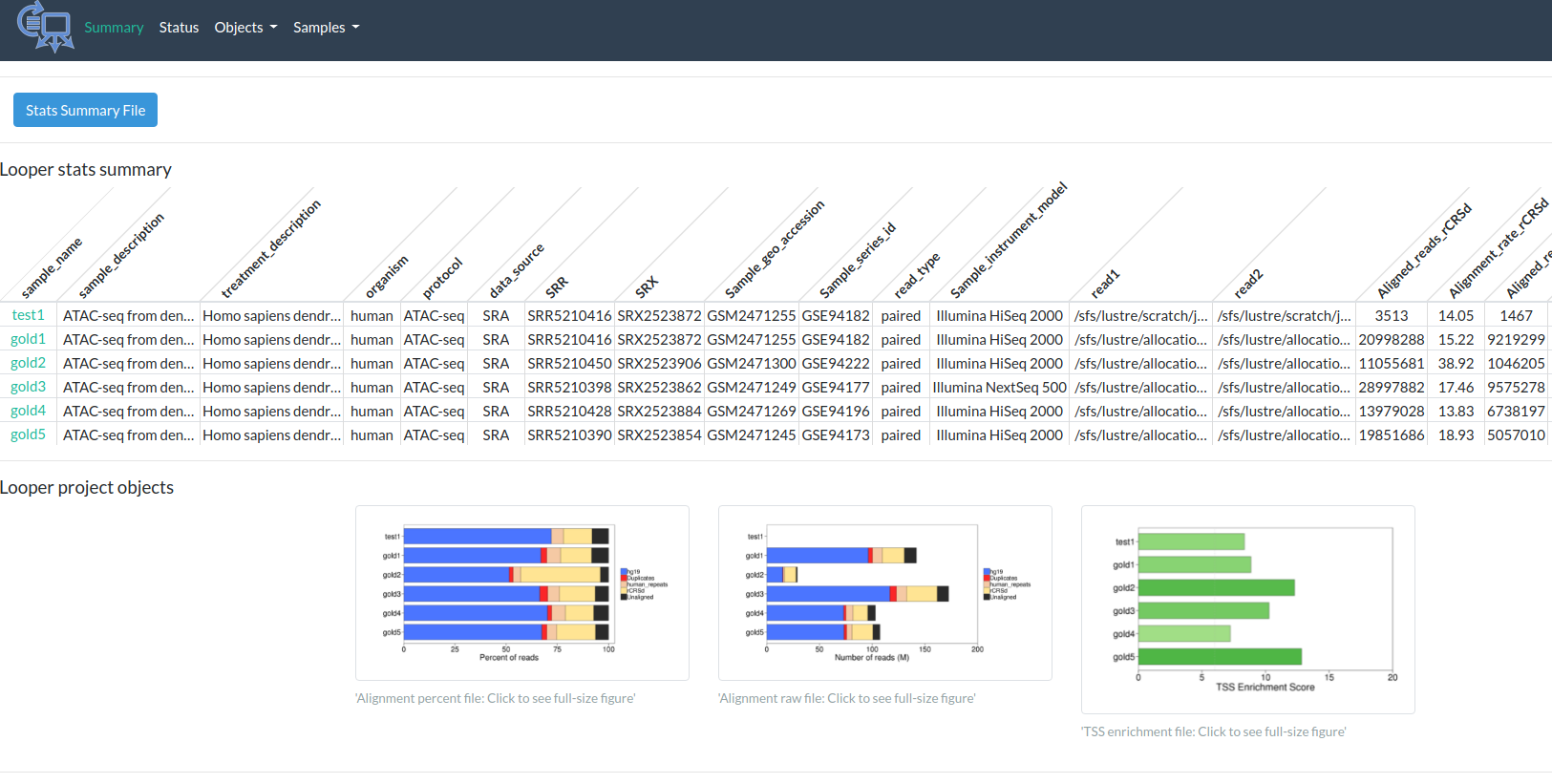
http://pepatac.databio.org/en/latest/files/examples/gold/gold_summary.html