Refgenie, PEP, and bulker
Nathan Sheffield, PhDA full-service reference genome manager.
The problem
Many tools require genome-related assets (like indexes).How should we organize these on disk?

Refgenie consists of 3 components


Refgenie splits tasks between CLI and server

The build/pull method needs provenance checks
Asset provenance:

Genome provenance:

Refget
Refget enables access to reference sequencesusing an identifier derived from the sequence itself.
How refget works

Refget v2.0: Collections for genome provenance

Recursive checksums have advantages
Allows getting content list only
Preserves chromosome order
Re-uses the checksum function
Duplicates are stored only once
Go one step further for...
Preserves chromosome order
Re-uses the checksum function
Duplicates are stored only once
Go one step further for...

It keeps going... and going...

Asset provenance:
Recipes + containers?
Genome provenance:
Solved by refget v2.0?
Tying human identifiers to a digest:
hg38:
refget_digest: 32a37a52a377d95bfd4b3d66763e1396a4480f34ab5c318a
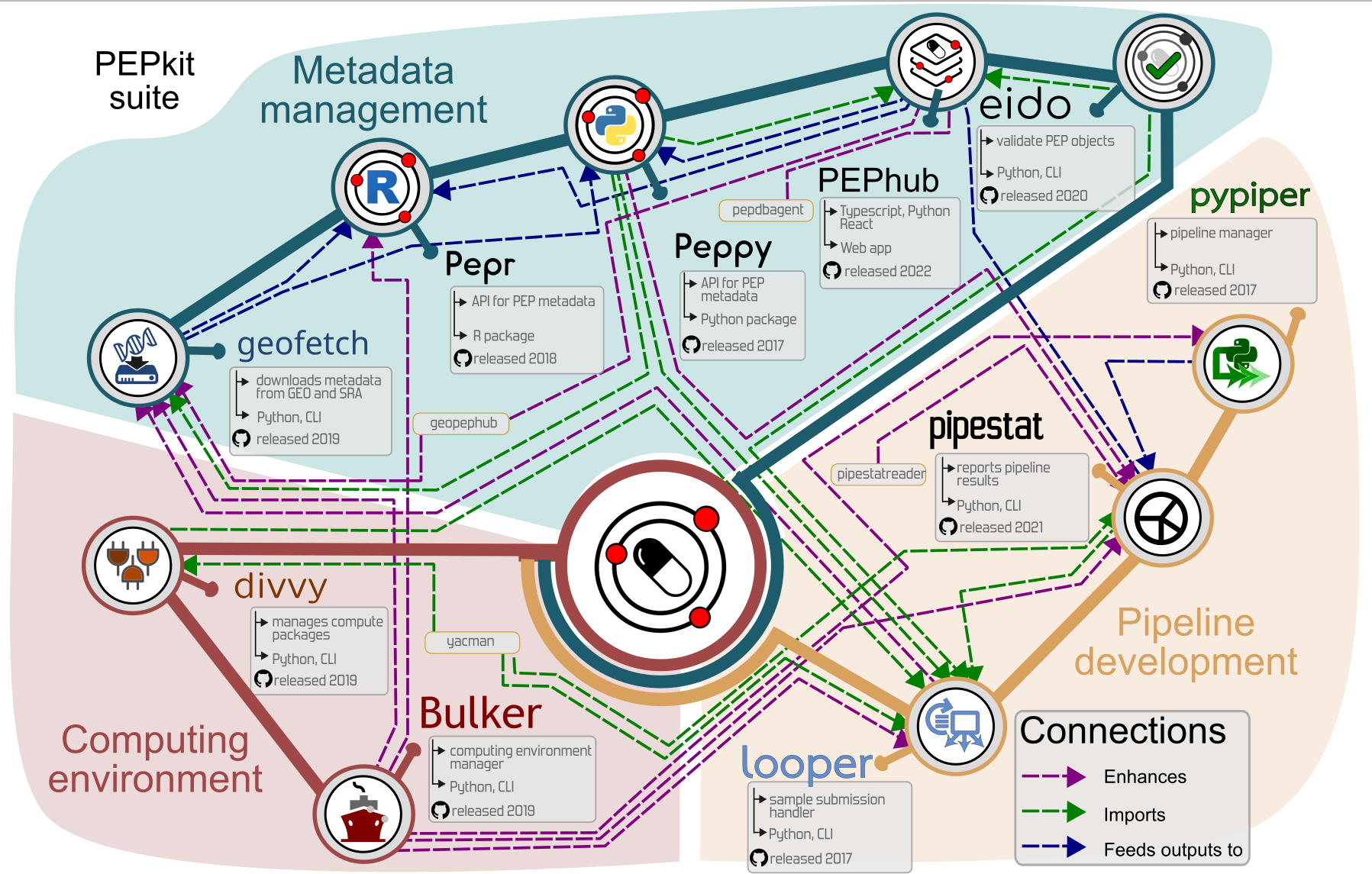
Pepkit
A structure and toolkit for organizing large-scale,sample-intensive biological research projects
Research is organized in projects


How do we conceptualize a research project?
Each project has 3 components

Organizing multiple projects is a challenge

How do I re-use a component?

A project is a set of edges in a tripartite graph

Enable linking with interfaces

We are building a modular ecosystem

pepkit · geofetch · looper · caravel · pypiper · divvy
PEP: Portable Encapsulated Projects

 PEP format
PEP format
sample_name,protocol,organism,input_file
frog_0h,RNA-seq,frog,/path/to/frog0.gz
frog_1h,RNA-seq,frog,/path/to/frog1.gz
frog_2h,RNA-seq,frog,/path/to/frog2.gz
frog_3h,RNA-seq,frog,/path/to/frog3.gz
PEP format
sample_name,protocol,organism,input_file
frog_0h,RNA-seq,frog,/path/to/frog0.gz
frog_1h,RNA-seq,frog,/path/to/frog1.gz
frog_2h,RNA-seq,frog,/path/to/frog2.gz
frog_3h,RNA-seq,frog,/path/to/frog3.gz
sample_table: /path/to/samples.csv
output_dir: /path/to/output/folder
other_variable: value
Add programmatic sample and project modifiers.
Automatically build new sample attributes from existing attributes.
Without derived attribute:
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | /path/to/frog0.gz |
| frog_1h | 1 | RNA-seq | frog | /path/to/frog1.gz |
| frog_2h | 2 | RNA-seq | frog | /path/to/frog2.gz |
| frog_3h | 3 | RNA-seq | frog | /path/to/frog3.gz |
Using derived attribute:
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | my_samples |
| frog_1h | 1 | RNA-seq | frog | my_samples |
| frog_2h | 2 | RNA-seq | frog | my_samples |
| frog_3h | 3 | RNA-seq | frog | my_samples |
| crab_0h | 0 | RNA-seq | crab | your_samples |
| crab_3h | 3 | RNA-seq | crab | your_samples |
| sample_name | t | protocol | organism | input_file |
| ------------- | ---- | :-------------: | -------- | ---------------------- |
| frog_0h | 0 | RNA-seq | frog | my_samples |
| frog_1h | 1 | RNA-seq | frog | my_samples |
| frog_2h | 2 | RNA-seq | frog | my_samples |
| frog_3h | 3 | RNA-seq | frog | my_samples |
| crab_0h | 0 | RNA-seq | crab | your_samples |
| crab_3h | 3 | RNA-seq | crab | your_samples |
Project config file:
sample_modifiers:
derive:
attributes: [input_file]
sources:
my_samples: "/path/to/my/samples/{organism}_{t}h.gz"
your_samples: "/path/to/your/samples/{organism}_{t}h.gz"Benefit: Enables distributed files, portability
Add new sample attributes conditioned on values of existing attributes
Before:
| sample_name | protocol | organism |
| ------------- | :-------------: | -------- |
| human_1 | RNA-seq | human |
| human_2 | RNA-seq | human |
| human_3 | RNA-seq | human |
| mouse_1 | RNA-seq | mouse |
After:
| sample_name | protocol | organism | genome |
| ------------- | :-------------: | -------- | ------ |
| human_1 | RNA-seq | human | hg38 |
| human_2 | RNA-seq | human | hg38 |
| human_3 | RNA-seq | human | hg38 |
| mouse_1 | RNA-seq | mouse | mm10 |
| sample_name | protocol | organism |
| ------------- | :-------------: | -------- |
| human_1 | RNA-seq | human |
| human_2 | RNA-seq | human |
| human_3 | RNA-seq | human |
| mouse_1 | RNA-seq | mouse |
Project config file:
sample_modifiers:
imply:
- if:
organism: human
then:
genome: hg38
- if:
organism: mouse
then:
genome: mm10Benefit: Divides project from sample metadata
Define activatable project attributes.
project_modifiers:
amendments:
diverse:
metadata:
sample_annotation: psa_rrbs_diverse.csv
cancer:
metadata:
sample_annotation: psa_rrbs_intracancer.csvBenefit: Defines multiple similar projects in a single file
Reproducibility
data + code+ environment
Containers
A promising solution, but how should we use them?
Combined

Individual

| easy to deploy |
| easy to use |
| reusable |
| combinable |
| subsetable |
| space efficient |
| Combined |
| Individual |
| Bulker |
How bulker does it
Two conceptual advances:
Containerized executables

Distribute containers in sets
