Introduction to Sequence Collections
Nathan Sheffield, PhDUnique identifiers and API for sequence collections.
Problem
Who is the authoritative provider of the reference genome?
- NCBI?
- UCSC?
- Ensembl?
- hard, soft, or no repeat masking?
- are alternative scaffolds included?
- are haplotypes included?
- how are chromosomes named (chr1, 1, or NC_000001.11)?
- how is the assembly named (hg38, GRCh38, or GCF_000001405.39)?
- Are any decoy sequences included (like EBV)?
| Provider | Chr1 name | Chr1 length | Chr1 md5 | Num chroms |
|---|---|---|---|---|
| Ensembl primary | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 195 |
| Ensembl toplevel | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 649 |
| NCBI | NC_000001.11 | 248956422 | 6aef897c3d6ff0c78aff06ac189178dd | 640 |
| UCSC | chr1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 456 |
https://gist.github.com/andrewyatz/692f81baab1bebaf09c481937f2ad6c6
Subtle differences in reference assembly lead to:
- Lack of reproducibility of analysis
- Lack of reusability of results
Solution

Refget -> Sequence collections
Refget
Refget enables access to reference sequencesusing an identifier derived from the sequence itself.
How refget works

Limitations
- only handles a single sequence
- excludes chromosome names
- no capacity for annotation
Extending to sequence collections
We need:- 1. An algorithm to create a deterministic, unique digest from a collection of sequences
- 2. A server capable of retrieving sequences given an identifier
First pass: Refgenie approach


Limitations and discussion
- Should we include sequence topology in the digest?
- What other attributes could we include?
- Are there better delimiters?
- How do we construct the 'string-to-digest'?
- How do we handle order of sequences?
- How should the API respond to requests?
Project goal:
The project specifies:
How do we digest a sequence collection?
JSON object: each sequence collection attribute is a property
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
],
"sequences": [
"31fc6ca291a32fb9df82b85e5f077e31",
"92c6a56c9e9459d8a42b96f7884710bc",
"5f63cfaa3ef61f88c9635fb9d18ec945"
]
}
← length of the sequences
← names of the sequences
← refget digests
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
],
"sequences": [
"31fc6ca291a32fb9df...",
"92c6a56c9e9459d8a4...",
"5f63cfaa3ef61f88c9..."
]
}
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
]
}
{
"lengths": [
4,
4,
8
],
"names": [
"chr1",
"chr2",
"chrX"
],
"sequences": [
"31fc6ca291a32fb9df...",
"92c6a56c9e9459d8a4...",
"5f63cfaa3ef61f88c9..."
],
"topologies" [
"linear",
"linear",
"circular"
]
}
Digest algorithm
- Canonicalize each attribute following RFC-8785 (JSON Canonicalization Scheme)
- Digest each string (GA4GH digest: SHA512 truncated to 24 bits, converted to base64)
- Canonicalize the entire object
- Digest the canonicalized string
 Tim Cezard
Tim Cezard
Advantages
- Accommodates new attributes with backwards-compatibility
- Additional layer of recursion to assess individual attributes
- Relies on existing JCS standard for string encoding
What gets digested?
Comparison function
| Provider | Chr1 name | Chr1 length | Chr1 md5 | Num chroms |
|---|---|---|---|---|
| Ensembl primary | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 195 |
| Ensembl toplevel | 1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 649 |
| NCBI | NC_000001.11 | 248956422 | 6aef897c3d6ff0c78aff06ac189178dd | 640 |
| UCSC | chr1 | 248956422 | 2648ae1bacce4ec4b6cf337dcae37816 | 456 |
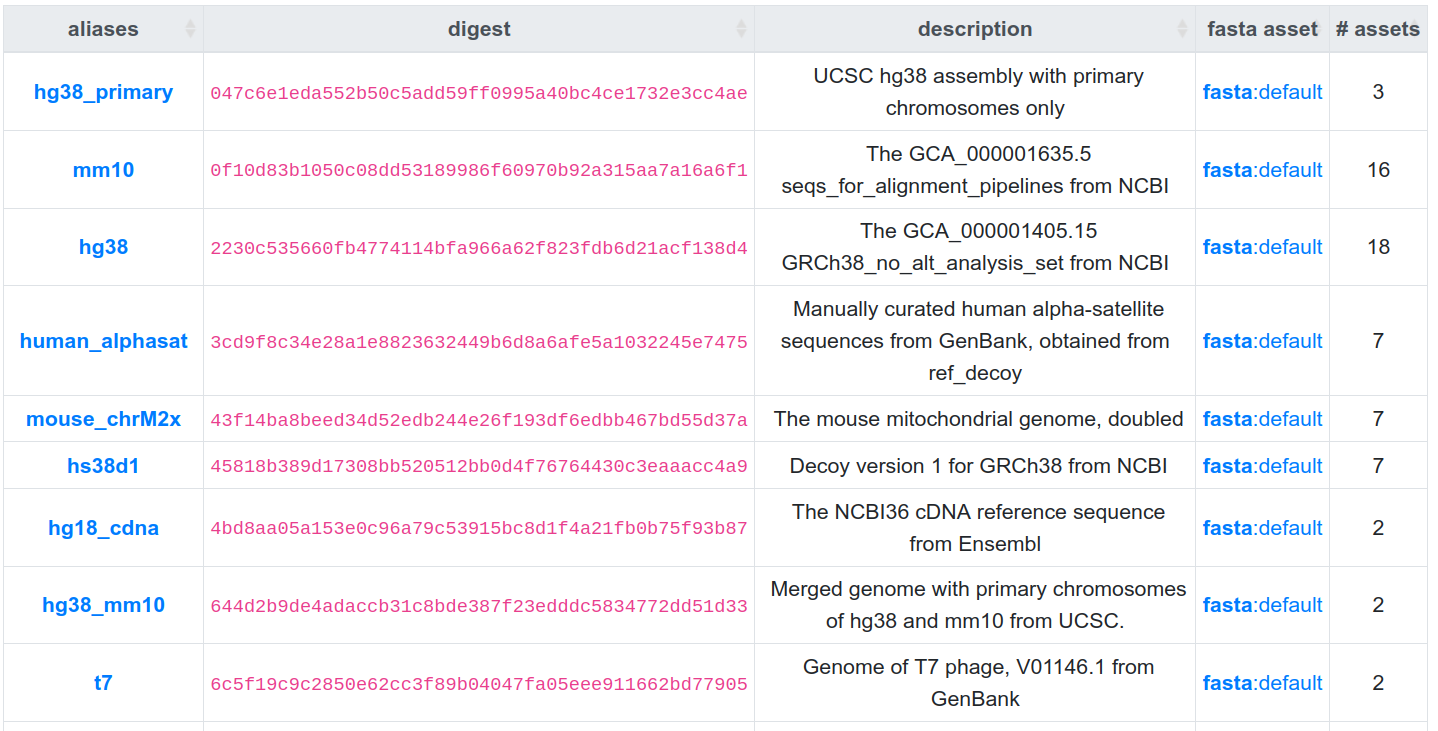
- seqcol 1: 047c6e1eda552b50c5add59ff0995
- seqcol 2: 2230c535660fb4774114bfa966a62
How compatible are they?
Comparison endpoint
{
"digests": {
"a": "59319772d1bcf2e0dd4b8a296f2d9682",
"b": "2e7bc302a54ecec62d8155e19fbf2748"
},
"arrays": {
"a-only": [],
"b-only": [],
"a-and-b": [
"lengths",
"names",
"sequences",
"names_lengths"
]
},
"elements": {
"total": {
"a": 3,
"b": 3
},
"a-and-b": {
"lengths": 3,
"names": 3,
"sequences": 3,
"names_lengths": 3
},
"a-and-b-same-order": {
"lengths": false,
"names": false,
"sequences": false,
"names_lengths": true
}
}
}
Seqcol API demonstration
https://seqcolapi.databio.org/API endpoints
GET /service-infoGET /collection/:digestGET /comparison/:digest1/:digest2POST /comparison/:digest1Conclusions
- Refget provides universal IDs for individual sequences
- Sequence collections extends this to reference genomes
- Using a deterministic algorithm, you can find the identifier
- A lookup service can retrieve the original sequence
- A comparison function allows fine-grained compatibility tests
- Please follow along: https://github.com/ga4gh/seqcol-spec